Toggle navigation
About Us
FAQ
Blog
Code of Ethics
Testimonials
More
Podcast
Directory
Login
Sign Up
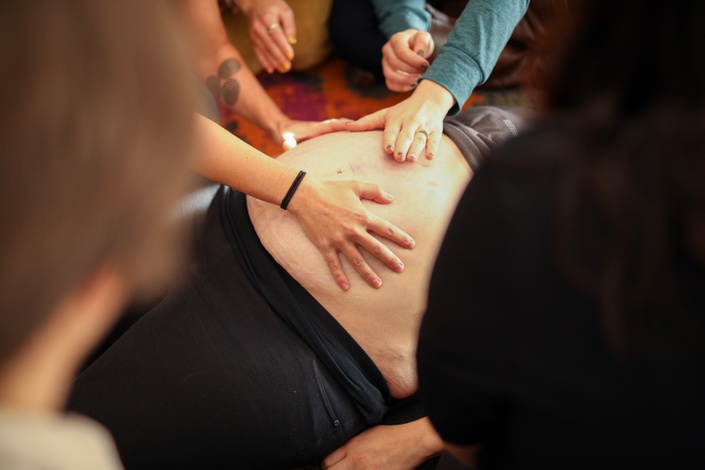
Educating Birth Workers & Women Everywhere!
Enroll Now
Choose Which Course Resonates With Your Goals
Holistic Birthkeeper & Doula Training
Available until
Teaching birth workers how to go above and beyond, to love, respect and support pregnant and birthing women in a holistic way!
HERBAL Training
%
COMPLETE
$555
Placenta Preparation Course
Available until
How to be 'the cake' maker of the birth world!
HERBAL Training
%
COMPLETE
$222
Peer Lactation Counselor
Available until
Train to work as a breastfeeding professional, educating and supporting mamas through breastfeeding successes and struggles.
HERBAL Training
%
COMPLETE
$333
Medical Setting Birth Support
Available until
NON-CERTIFYING COURSE- FOR INFO PURPOSES ONLY: Hospital, Birth Center, and Midwife Birth Support, and more. Guidance on how to best support in these settings.
HERBAL Training
%
COMPLETE
$111
Womb Reading: A Holistic Fertility Guide
Available until
Everything you need to know about monitoring, tracking, documenting, and evaluating your fertility.
HERBAL Training
%
COMPLETE
$111
Freebirth Course
Available until
Discussing the basics in safety and need-to-knows in an unassisted birth setting for expecting moms for happy unassisted birthing!
HERBAL Training
%
COMPLETE
$222
The Whole SHABANG Course Bundle
Available until
ALL Of Our Currently Available Courses Bundled!
12 Course Bundle
%
COMPLETE
$999
Not So Bloody Course Bundle
Available until
A Trio Bundle of Doula & Birthkeeper, Peer Lactation Counselor, & Holistic Fertility Certification Courses
3 Course Bundle
%
COMPLETE
$700
The Trio Course Bundle
Available until
A bundle of the Doula & Birthkeeper, Peer Lactation Counselor, and Placenta Specialist courses
3 Course Bundle
%
COMPLETE
$777
Travel Attending
Available until
Taylor
%
COMPLETE
FREE